전문약 품목허가 신청시 ‘기준 및 시험방법에 관한 자료’-‘생물학적 동등성시험' 자료 의무화
 |
앞으로 이미 허가받은 품목과 같은 제조소에서 동일한 제조공정 중 제조설비·단위 및 포장·용기가 다른 공정으로 위탁 생산되는 전문약은 3개 제조단위의 GMP 자료를 제출해야 한다.
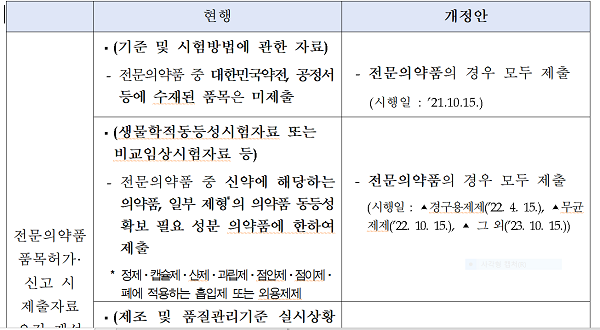
또 전문약 품목허가 신청시 ‘기준 및 시험방법에 관한 자료’와 ‘생물학적 동등성시험 등에 관한 자료’를 제출하는 쪽으로 품질관리가 강화된다.
시행일은 경구용제제는 오는 2022년4월15일▴무균제제는 2022년10월15일▴그 외는 2023년10월15일부터다.
식품의약품안전처(처장 이의경)는 의약품의 품질·안전관리 기준을 강화하고, 임상시험계획 변경 절차를 완화하는 것을 주요 내용으로 하는 '의약품 등의 안전에 관한 규칙'(총리령)을 10월 14일 개정·공포했다고 밝혔다.
이번에 개정한 총리령의 주요 내용은 전문약의 품질·안전관리 강화를 위하여 ▲기준 및 시험방법 ▲생물학적동등성시험 ▲의약품 제조 및 품질관리 기준(GMP) 등의 심사·평가 제도를 미국·유럽 등 국제적인 수준으로 개선된다.
구체적으로는 모든 전문약에 대해 품목허가를 신청하는 경우 ‘기준 및 시험방법에 관한 자료’와 ‘생물학적 동등성시험 등에 관한 자료’ 등을 제출받아 품질관리가 강화된다.
또 기존에 허가받은 품목과 같은 제조소에서 동일한 제조공정으로 위탁생산하는 전문약의 경우 지금까지는 ‘의약품 제조 및 품질관리기준(GMP)’ 자료 제출이 면제됐으나, 앞으로는 3개 제조단위에 대한 자료를 제출하는 쪽으로 강화된다.
제조공정뿐만 아니라 제조설비, 제조단위, 포장·용기까지 동일한 경우에는 1개 제조단위 자료 제출해 왔으나 앞으로는 다른 경우 3개 제조단위 자료를 제출하게 되는 것이다.
식약처는 업계 애로사항을 해소하기 위해 절차적 규제도 개선·보완한다.
임상시험계획의 ‘변경승인’ 대상이었던 ▲시험군·대조군 추가, ▲임상시험 종료기준 변경, ▲투약방법의 변경 등을 ‘변경보고’ 대상으로 전환해 신속하고 원활하게 임상시험이 진행될 수 있도록 개선된다.
또한 ‘마약류통합관리시스템’을 통해 제조·수출·수입 현황을 별도로 보고하는 마약 및 향정신성약의 경우에는 '의약품 등의 안전에 관한 규칙'에 규정된 의약품 생산·수입실적 보고 대상에서 제외하도록 규제가 정비된다.
이어 의약품 품목허가·신고시 ‘안정성에 관한 자료’만을 검토할 경우 처리기한을 단축해 신속하게 허가할 수 있는 기반이 마련된다.
각각 허가는 70일→45일, 신고는 55일→30일, 변경허가는 65일→40일로 단축되는 셈이다.
식약처는 이번 개정을 통해 의약품 개발·허가 절차를 국제기준에 맞춰 합리적으로 개선함으로써 의약품 전반에 대하여 품질 향상을 유도하는 한편 신속한 의약품 출시를 지원할 수 있는 환경을 조성할 것으로 기대한다고 밝혔다.
한정렬 기자 jrh05@hanmail.net
<저작권자 © 데일리메디팜, 무단 전재 및 재배포 금지>
 반복해서 떠오르는 생각(강박사고)이 일상을 방해한다면(?)
반복해서 떠오르는 생각(강박사고)이 일상을 방해한다면(?)
