비임상·임상 자료 추가 검토-품질자료 심사 중
아스트라제네카 코로나19 백신 안전·효과성 검증 자문단’ 회의 31일에 실시...2월 1일 결과 공개
추가자료의 예방효과‧신청 용법‧용량 타당성‧안전성 등 검토후 국가출하승인 위한 품질분야 심사에 집중
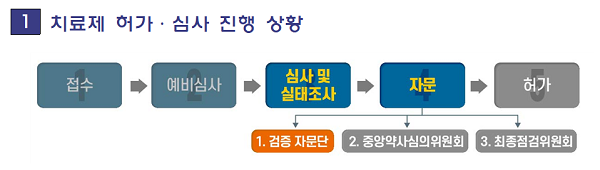 |
식약처는 22일 ㈜셀트리온이 허가 신청한 코로나 항체치료약 ‘렉키로나주’와 관련해 "제조소 및 임상시험실시 의료기관에 대한 실태조사를 마쳤다"며, "현재 비임상·임상시험 자료에 대해 추가 검토와 품질자료에 대한 심사를 진행하고 있다"고 밝혔다.
품질자료는 치료제의 제조공정관리, 품질의 적합성 등에 관한 자료로서, 원료약 및 완제약의 물리화학적·생물학적 성질, 제조법, 기준 및 시험방법, 표준품, 용기·포장 등에 관한 자료를 심사하고 있다는게 식약처의 설명이다.
또 유전자재조합약의 특성을 고려한 원료관리 자료, 공정단계별 불순물 제거 검증 자료, 보관방법 및 사용기간 설정을 위한 안정성시험 자료를 검토하고 있다.
식약처는 임상시험실시 의료기관 실태조사에 대해 "임상시험 참여 환자의 안전과 시험결과의 신뢰성을 확인하기 위해 환자 동의서 작성, 임상시험용의약품 관리, 전자 자료 관리 등 임상시험 전반에 걸쳐 규정 준수 여부를 서류검토와 현장 점검(1월12일~15일)을 통해 확인했다"며 "해외 의료기관의 경우 임상시험의 신뢰성과 국제적 윤리기준에 따라 수행되었는지 서류상으로 중점 검토했다"고 말했다.
식약처는 허가·심사 관련 향후 계획에 대해 "㈜셀트리온 ‘렉키로나주’와 관련해 아직 제출되지 않은 품질자료 일부 등에 대한 자료 제출을 1월20일 요청했으며, 제출되는 대로 심사를 계속 진행할 예정"이라고 밝혔다.
이어 "심사 결과를 종합해 식약처의 법정 자문기구인 ‘중앙약사심의위원회’에서 신청 품목의 안전성, 효과성, 허가시 고려해야 할 사항 등에 대해 오는 27일에 자문받고, 그 결과를 당일 공개할 예정"이라고 밝혔다.
아스트라제네카社 ‘아스트라제네카 코비드-19백신주’와 관련 "비임상, 임상, 품질(미제출자료) 등 심사 자료를 추가로 1월15일 요청했으며, 자료가 제출되는 대로 예방효과, 신청한 용법‧용량의 타당성, 안전성 등에 대해 검토하고, 국가출하승인을 위한 품질분야 심사에 집중할 예정"이라며 "임상시험 자료와 관련해 외부 전문가들이 참여하는 ‘코로나19 백신 안전·효과성 검증 자문단’ 회의를 1월 31일에 실시하고, 그 결과를 2월 1일 공개할 예정"이라고 설명했다.
자문단은 감염내과 중심의 임상전문가, 비임상, 품질분야에 전문지식과 경험이 풍부한 외부 전문가로 구성되며, 신청품목의 안전성과 효과성, 임상적 의의, 대상환자의 적정성 등에 대하여 자문하게 된다.
▶식약처, ‘아스트라제네카 코비드-19백신주’와 관련 제조소 'SK바이오사이언스' 실태조사 마쳐
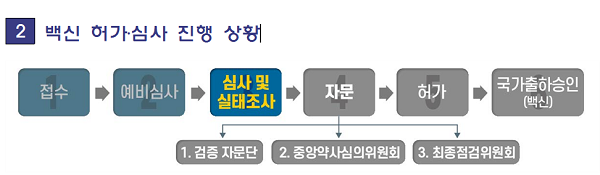
아울러 식약처는 ㈜한국아스트라제네카가 허가 신청한 ‘아스트라제네카 코비드-19백신주’와 관련해 "제조소 'SK바이오사이언스'에 대한 실태조사를 마쳤으며, 현재 비임상·임상시험 자료와 품질자료에 대한 심사를 진행하고 있다"고 밝혔다.
백신의 예방효과 확인은 임상시험 참여자 중 코로나19 감염환자가 목표 수에 도달하면 백신접종군(시험군)과 위약접종군(대조군)의 감염환자 비율로 예방효과를 분석·평가한다.
WHO에 따르면 코로나19 백신의 예방효과 50% 이상으로 권고하고 있다.
이밖에 백신접종 후 인체에 생성되는 다양한 항체와 그 양을 측정해 면역반응이 일어난 정도를 평가할 수 있는데, 그 지표로는 코로나19 바이러스와 결합하는 ‘결합항체가’, 바이러스를 무력화시키는 ‘중화항체가’ 등이 있다.
식약처는 또 "백신접종군과 위약접종군 간 코로나19 중증환자 발생율을 비교해 백신 접종에 따라 중증환자의 발생을 얼마나 줄일 수 있는지 예측할 수 있다"며 "중증환자 발생을 줄인다면 코로나19에 따른 사망자를 감소시키고 의료시스템 부족에도 도움을 줄 수 있다"고 말했다.
식약처는 "백신의 안전성은 영국(2건), 브라질, 남아프리카공화국에서 수행된 4건의 임상시험 결과를 통합해 평가하고 있다"며 "평가법은 예측되거나 예측되지 않은 이상사례 발생에 대해 백신접종군과 위약접종군을 동시에 모니터링해 비교 평가하며, 접종 후 28일 이내의 모든 이상사례를 검토한다. 예측되는 이상사례는 주사부위 통증, 오심, 발열, 두통, 근육통, 관절통 등"이라고 설명했다.
장기 안전성 추적조사는 투여 후 1년 동안 진행한다.
 |
또한 사망사례를 비롯해 아나필락시스와 같은 생명을 위협하거나 중대한 이상사례를 중점적으로 검토하고 이상사례와 백신과의 인과관계를 평가하여 인과관계가 있다고 판단되는 경우 위해성관리계획의 안전성 중점검토항목으로의 관리 여부를 고려하게 된다.
위해성관리계획(Risk Management Plan, RMP)는 의약품의 허가 전과 시판후 전 주기에 걸쳐 안전・효과성에 관한 정보수집, 조사, 시험, 위해성 발생 최소화 등을 위해 실시하는 종합 안전관리 활동계획이다.
식약처는 "65세 이상 고령자에게서 특별히 주의가 요구되는 안전성 정보가 있는지 백신접종군과 위약접종군을 비교·검토하고 있다"며 "영국의 긴급사용 승인시 평가된 4건의 임상시험에서의 만 65세 이상 고령자의 비율은 안전성평가 대상자의 약 10%며 비임상·임상시험 자료 등을 종합적으로 검토해 허가사항의 사용상 주의사항에 반영할 계획"이라고 설명했다.
사용상의 주의사항은 경고, 금기, 신중투여, 이상반응, 일반적 주의, 약물상호작용, 임부·수유부·고령자 등 투여시 주의사항, 과량투여시 처치, 보관상 주의사항 등이다.
식약처는 품질자료는 제품의 제조공정관리, 품질관리 등에 관한 자료로써, 원료약 및 완제약의 물리화학적·생물학적 성질, 제조방법, 기준 및 시험방법, 표준품, 용기·포장 등에 관한 자료를 심사하고 있다.
또한 공정단계별 불순물 제거 검증 자료, 보관방법 및 사용기간 설정을 위한 안정성시험 자료를 검토하고 있다.
▶식약처, SK바이오사이언스㈜ 제조‧품질관리기준(GMP) 실시 상황 평가
식약처는 허가신청 제품이 일관된 품질로 생산될 수 있는 시설과 품질보증체계 등을 갖추었는지 평가하기 위해 1월18일~20일 식약처 조사팀이 SK바이오사이언스㈜ 제조소에 대한 현장 실태조사를 실시했다고 밝혔다.
이번 허가신청 제품은 바이러스벡터 백신으로 일반적인 백신과 달리 바이러스(전달체 역할)를 사용하므로 제조소 내 유전자변형생물체 관리, 생물안전등급(BSL) 관리에 관한 사항 등을 추가적으로 확인했다.
바이러스벡터 백신은 전달체로 사용하는 다른 바이러스 유전자에 감염병을 일으키는 바이러스 항원 유전자를 삽입해서 대량 생산하는 백신이다.
식약처는 또한 "해당 백신은 무균 주사제로서 제조하는 구역이 미세입자나 미생물로부터 오염을 방지할 수 있는 시설·환경을 갖추고 정해진 청정수준을 유지하고 있는지 현장 실태조사를 통해 확인했다"며 "이미 생산된 완제약과 원료단계인 원액에 대한 실제 제조기록 및 품질검사기록을 확인하고 데이터 신뢰성 등에 대해 검토했다"고 밝혔다.
국내 백신 제조업체에 대한 GMP 평가와 관리역량은 세계보건기구(WHO)의 품질인증(PQ) 등을 통해 국제적으로 인정받고 있다.
WHO PQ(Pre-Qualification)는 WHO가 국제조달을 통해 개발도상국 공급을 목적으로 의약품의 품질 및 안전성·유효성을 평가하는 제도로, 생산국 규제당국의 안전관리 역량을 포함해 평가된다.
현재 국내 5개 업체 19개 품목이 세계보건기구(WHO)로부터 품질인증(PQ)을 받아 유엔아동기금(유니세프) 등 UN 산하기관에 백신을 국제조달하고 있으며 2016년에는 식약처-세계보건기구(WHO) 간 업무협약을 맺고 식약처가 제공한 GMP 실사 보고서를 근거로 WHO 현장 조사를 대체하고 있다.
식약처는 "앞으로도 코로나19 치료제·백신의 안전성과 효과를 과학적으로 철저히 검증하는 한편, 다양한 전문가 의견을 수렴해 객관성과 투명성을 확보할 수 있도록 최선을 다하겠다"고 밝혔다.
한정렬 기자 jrh05@hanmail.net
<저작권자 © 데일리메디팜, 무단 전재 및 재배포 금지>
 반복해서 떠오르는 생각(강박사고)이 일상을 방해한다면(?)
반복해서 떠오르는 생각(강박사고)이 일상을 방해한다면(?)
